Research
1. 増殖シグナルはユビキチン化によって緻密に制御されています
Missionで説明したように、動物細胞の増殖は増殖因子によって適切に制御されています。増殖因子が細胞膜上の受容体に結合すると、受容体が活性化し、細胞内で増殖シグナルが発信されます。このシグナルに応答して細胞周期が進行し、DNAの複製や細胞分裂が起こります。増殖シグナルが過剰に伝達されると、腫瘍やがんの形成につながります。正常な細胞は、様々なしくみで過剰なシグナル伝達を防いでいます。例えば、“リガンド依存性受容体エンドサイトーシス”というしくみがあります。増殖因子(リガンド)の結合によって活性化した受容体が細胞内の小胞に取り込まれ(図1)、エンドソームを経て、最終的にリソソーム内の加水分解酵素によって分解されるしくみです。このしくみによって細胞膜上の受容体が少なくなり、増殖シグナルがある程度弱められます。研究室の立ち上げ当初、私たちは、リガンド依存性受容体エンドサイトーシスに着目して研究をはじめました。

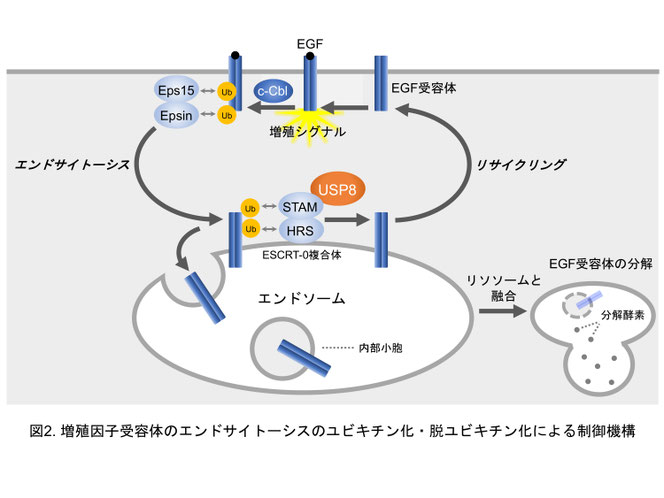
受容体エンドサイトーシスは、受容体の「ユビキチン化」が引き金となって起こることがわかっています(図2)。ユビキチン化は、ユビキチンという小さなタンパク質が共有結合する反応で、タンパク質の翻訳後修飾の一つです。一般に、標的タンパク質にユビキチンを連結させる酵素を「ユビキチンリガーゼ」と呼びます。増殖因子受容体に対してはc-Cblなどのユビキチンリガーゼが働きます。次に、Eps15やEpsinという「ユビキチン結合タンパク質」がユビキチン化された受容体を認識します。そして、これらの働きによって、細胞膜の細胞質側への陥入がはじまります。こうして細胞内にできた小胞は互いに融合してエンドソームになります。次に、エンドソーム上の受容体は、ユビキチン結合タンパク質であるHRSとSTAMの複合体(ESCRT-0複合体と呼びます)によって認識されます。私たちは、このESCRT-0複合体の機能を詳しく調べました。その結果、ESCRT-0複合体は受容体をエンドソームの内部小胞に運ぶプロセスに関与していることがわかりました(Komada & Kitamura, Mol. Cell. Biol. 1995; Komada et al., J. Biol. Chem. 1997; Komada & Soriano, Genes Dev.1999; Mizuno et al., Mol. Biol. Cell 2003; Morino et al., Exp. Cell Res. 2004; Mizuno et al., J. Biochem. 2004; Komada & Kitamura, J. Biochem. 2005)。内部小胞の受容体は、最終的に、エンドソームがリソソームと融合することによって分解されます。
標的タンパク質からユビキチンを取り除く酵素を「脱ユビキチン化酵素」と呼びます。私たちは、脱ユビキチン化酵素USP8がESCRT-0複合体に結合していることを見つけました。そして、USP8がエンドソームで受容体を脱ユビキチン化し、受容体の細胞膜へのリサイクリングを促すことを明らかにしました(Mizuno et al., Mol. Biol. Cell 2005; Mizuno et al., Traffic 2006; Komada, Curr. Drug Discov. Technol. 2008; Mukai et al., EMBO J. 2010)。つまり、受容体エンドサイトーシスはユビキチン化と脱ユビキチン化のバランスによって調節されていることを明らかにしました(図2)。
2. ユビキチン化の異常は腫瘍性疾患の発症につながります
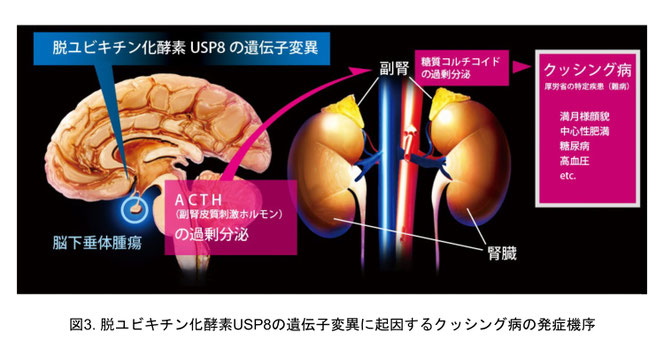
当時、私たちは、この制御に異常が生じると過剰な増殖シグナルが伝達されて腫瘍やがんの形成につながると予想していました。その後、実際に、USP8の遺伝子変異がある種の腫瘍で生じていることが発見されました。その腫瘍はクッシング病という難病を引き起こす下垂体腫瘍で、この腫瘍におけるUSP8の変異率は40%を超えます。
クッシング病は、下垂体腫瘍から副腎皮質刺激ホルモン(ACTH)が過剰に分泌され、これに応じて副腎から糖質コルチコイドが過剰に分泌される病気です。過剰な糖質コルチコイドによって満月様顔貌・中心性肥満・糖尿病・高血圧などの症状が起こり、未治療患者の5年生存率は約50%です(図3)。現在のところ、高度な技術を要する下垂体腫瘍の外科的切除が主な治療法となっており、特効薬はありません。
私たちは、USP8の遺伝子変異を見つけたドイツのグループと共同研究を進めました。その結果、遺伝子変異によってUSP8が過剰に活性化し、増殖シグナルが過剰になることを明らかにしました(Reincke et al., Nat. Genet. 2015; Perez-Rivas et al., J. Clin. Endocrinol. Metab. 2015; Theodoropoulou et al., Eur. J. Endocrinol. 2015; Hayashi et al., Eur. J. Endocrinol. 2016)。さらに、この腫瘍の特徴であるACTHの分泌異常についてもメカニズムを明らかにしつつあります。クッシング病の発症機構を分子レベルで理解し、その知見をもとに世界初の治療薬を開発する研究もスタートしています。基礎研究の成果を臨床に橋渡しすべく、使命感をもって取り組んでいます。
3. 多彩な細胞機能の制御にユビキチン化が関わることもわかってきました
ユビキチン化反応は、標的タンパク質にユビキチンがペプチド結合で連結する反応です。詳しく言うと、標的タンパク質のリジン残基(あるいはN末端メチオニン残基)のアミノ基に、ユビキチンのC末端のカルボキシル基が連結します。多くの場合、標的タンパク質に結合したユビキチンが新たな標的となって、そこに別のユビキチンが連結する反応が起こります。これが繰り返されて「ユビキチン鎖」が形成されるのが、この反応の特徴です。さらに話は複雑なのですが、ユビキチンに含まれる7カ所のリジン残基(あるいはN末端メチオニン残基)はいずれも連結部位として利用できます。従って、原理的には、8種類のつながり方のユビキチン鎖ができます。実際には、標的タンパク質ごとに関与するユビキチンリガーゼの種類が違っていて、この種類の違いに応じて作られるユビキチン鎖の連結部位が変わるようです。このような理由から、細胞内で形成されるユビキチン鎖は含まれるユビキチンの分子数や連結部位が様々で、立体構造に多様性があることがわかっています。
初期の研究によって、ユビキチン化には標的タンパク質の分解を誘導する働きがあることが明らかにされました。この研究には、ノーベル賞が与えられています。その後、ユビキチン化の他の働きも次々と明らかにされてきています。現在では、「異なる構造のユビキチン鎖が異なる種類のユビキチン結合タンパク質によって認識される結果、標的タンパク質が異なる制御を受ける」ことが明らかになりつつあります。
私たちは、受容体エンドサイトーシスの研究をきっかけにユビキチンの研究分野に参入しました。そして、様々なユビキチンリガーゼ・ユビキチン結合タンパク質・脱ユビキチン化酵素の性質を調べてきました。その結果、ユビキチン化が多彩な細胞機能の制御に関わることを明らかにしています。例えば、インスリンは血糖調節に重要なホルモンですが、インスリンの細胞内シグナル伝達はユビキチン化によって調節されます。詳しく言うと、シグナル伝達に重要な複合体の形成が、これを構成するタンパク質のユビキチン化によって促進されます(→福嶋による研究紹介の動画)(Fukushima et al., Nat.Commun. 2015; Fukushima et al., BBRC 2017)。細胞には、熱などのストレスに適応するためのストレス顆粒や、リボソームの合成に重要な核小体という細胞内小器官があります。これらの小器官の形成もユビキチン化によって調節されます(Endo et al., J. Cell Sci. 2009; Endo et al., J. Biol. Chem. 2009; Xie et al., J. Cell Sci. 2018)。生体内で最も多量に作られるタンパク質であるコラーゲンの細胞内輸送も、ユビキチン化によって調節されます(Kawaguchi et al., BBRC 2018)。このように、ユビキチン化は種々の細胞内構造物の形成や輸送を制御することによって、多彩な細胞機能の制御に関わることを明らかにしてきました(図4)。現在は、ホルモン分泌顆粒やウイルス粒子などに着目し、これらの形成や輸送におけるユビキチン化の役割を研究しています。

4. 胎盤や乳腺という哺乳類特有の組織で働く新しい増殖抑制タンパク質を発見しました
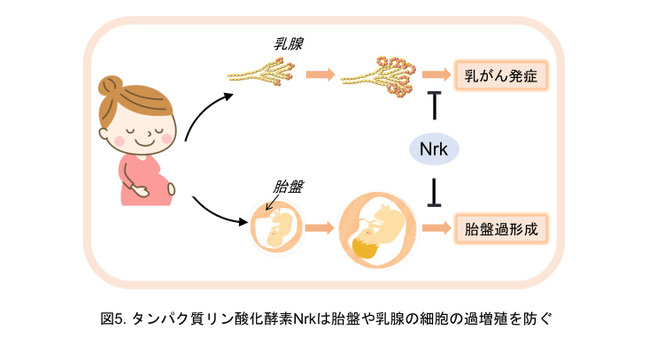
私たちは、増殖シグナルの制御機構を明らかにする研究の一環として、機能未知のタンパク質リン酸化酵素の解析も行っています。研究対象としたリン酸化酵素の一つが、Nik-related kinase(Nrk)です。Nrkは、胎盤や乳腺という哺乳類特有の組織に限定的に発現していることがわかりました。Nrkの遺伝子欠損マウスを作製したところ、胎盤の過形成が見られました。また、高頻度で乳がんが発症することもわかりました。これらのことから、Nrkは胎盤細胞や乳腺細胞の過増殖を防ぐ役割があると考えられます(Denda et al., J.Biol.Chem. 2011; Yanagawa et al., Am. J. Pathol. 2016; Naito et al., FEBS let. 2020)(図5)。現在、Nrkが増殖シグナルを抑制する詳細な分子メカニズムを明らかにしつつあります。さらに、Nrkの分子進化についても調べました。その結果から、Nrkは脊椎動物に広く存在していますが、哺乳類誕生の過程でNrkが急速に分子進化し、増殖シグナルを抑制する機能を獲得した可能性が示されています。
胎盤は妊娠期、乳腺は授乳期にそれぞれ急速に発達します。急速に発達するこれらの組織の細胞増殖を適切に制御するために、哺乳類専用の増殖制御タンパク質としてNrkが進化したとも考えられます。マウスのNrkは乳がんの発症を抑制しますが、今後、ヒトのNrkも同じかを検討する必要があります。ヒトでも乳がんの発症を抑制するならば、将来的には、Nrkの作用を模倣する物質を開発するなどにより、乳がんの治療に貢献できるかもしれません。そのような応用を見据えて、研究を進めたいと考えています。
5. 今後の研究
増殖シグナルは様々な状況に応じて変動します。細胞が置かれている環境が増殖にとって適していない場合は、増殖シグナルは伝達されません。例えば、栄養不足などのストレスを受けている状況では、増殖シグナルが伝達されにくくなることが知られています。
増殖シグナルは、細胞を含む組織がどのような発達段階・生理状態にあるかによっても強く影響されます。例えば、授乳期には、種々のホルモンが乳腺細胞に働きかけて増殖シグナルを強め、乳腺が発達します。授乳期を過ぎるとこのような働きかけがなくなり、増殖シグナルは弱まって、乳腺は退縮します。このようなしくみによって、適切なタイミングで適切な部位の細胞増殖が起こることが可能になっています。
長いタイムスケールで見てみると、生物進化の過程で増殖シグナルの制御システムも進化しており、それぞれの生物に適した制御が行われるよう最適化されているようです。哺乳類になる過程で起きたNrkの分子進化は、その好例と言えます。
このように、動物細胞の増殖シグナルは、大小の時空間スケールにわたって様々な制御を受けています。しかし、どのような分子機構で制御されているか、またその意義とは何か、まだ多くの謎が残されています。私たちは、細胞生物学・生化学・分子生物学の研究手法に加えて、最新のプロテオミクス・細胞イメージング技術・バイオインフォマティクスなどを用いて、その謎に挑んでいます。
現在の研究内容について詳しく聞きたい大学院受験生の方は、福嶋 (tofu [at] bio.titech.ac.jp)までお問い合わせください。